![]()



![]()
Needleman-Wunsch and Smith-Waterman algorithms in python for any iterable objects.
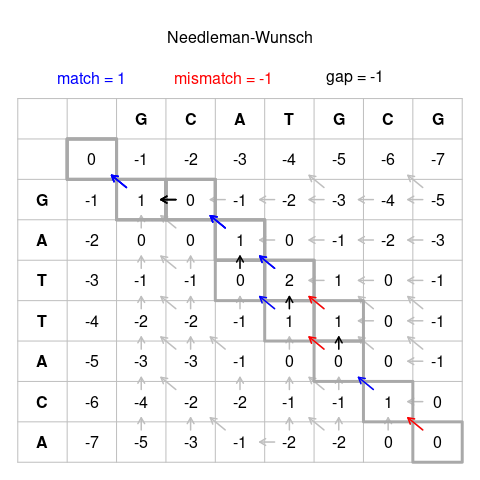
The Needleman–Wunsch algorithm is an algorithm used in bioinformatics to align protein or nucleotide sequences. It was one of the first applications of dynamic programming to compare biological sequences. The algorithm was developed by Saul B. Needleman and Christian D. Wunsch and published in 1970. The algorithm essentially divides a large problem (e.g. the full sequence) into a series of smaller problems and uses the solutions to the smaller problems to reconstruct a solution to the larger problem. It is also sometimes referred to as the optimal matching algorithm and the global alignment technique. The Needleman–Wunsch algorithm is still widely used for optimal global alignment, particularly when the quality of the global alignment is of the utmost importance.
-- From the Wikipedia article

The Smith–Waterman algorithm performs local sequence alignment; that is, for determining similar regions between two strings of nucleic acid sequences or protein sequences. Instead of looking at the entire sequence, the Smith–Waterman algorithm compares segments of all possible lengths and optimizes the similarity measure.
-- From the Wikipedia article
from minineedle import needle, smith, core
# Use miniseq objects
# Load sequences as miniseq FASTA object
import miniseq
fasta = miniseq.FASTA(filename="myfasta.fa")
seq1, seq2 = fasta[0], fasta[1]
# Or use strings, lists, etc
# seq1, seq2 = "ACTG", "ATCTG"
# seq1, seq2 = ["A","C","T","G"], ["A","T","C","T","G"]
# Create the instance
alignment: needle.NeedlemanWunsch[str] = needle.NeedlemanWunsch(seq1, seq2)
# or
# alignment smith.SmithWaterman[str] = smith.SmithWaterman(seq1, seq2)
# Make the alignment
alignment.align()
# Get the score
alignment.get_score()
# Get the sequences aligned as lists
al1, al2 = alignment.get_aligned_sequences(core.AlignmentFormat.list) # or "list"
# Get the sequences as strings
al1, al2 = alignment.get_aligned_sequences(core.AlignmentFormat.str) # or "str
# Change the matrix and run again
alignment.change_matrix(core.ScoreMatrix(match=4, miss=-4, gap=-2))
alignment.align()
# Print the sequences aligned
print(alignment)
# Change gap character
alignment.gap_character = "-gap-"
print(alignment)
# Sort a list of alignments by score
first_al = needle.NeedlemanWunsch(seq1, seq2)
second_al = needle.NeedlemanWunsch(seq3, seq4)
for align in sorted([first_al, second_al], reverse=True):
print(align)pip install minineedleNeedleman-Wunsch alignment class. It has the following attributes:
- seq1
- seq2
- alseq1
- alseq2
- nmatrix
- pmatrix
- smatrix
- score
- identity
- gap_character
To create the instance you have to provide two iterable objects with elements that can be compared with "==".
Smith-Waterman alignment class. It has the following attributes:
- seq1
- seq2
- alseq1
- alseq2
- nmatrix
- pmatrix
- smatrix
- score
- identity
To create the instance you have to provide two iterable objects with elements that can be compared with "==".
With this class you can define your own score matrices. It has three attributes:
- match
- miss
- gap
Performs the alignment.
Returns the score of the alignment. It runs align() if it has not been done yet.
Takes a ScoreMatrix object and updates the matrix for the alignment. You still have to run it calling align().
Returns the % of identity (rounded with 2 decimal points).
Return the alignment matrix as a list of lists.