An SVM-based classifier to predict genome enhancers based on ChIP-Seq and ATAC-Seq signals.
This is my final project in "Methods in Bioinformatics" Course of Prof. Ge Gao in the autumn semaster of 2019. Here I designed a novel classifier to predict genome enhancers. The imput features include chromatin modification and related enzymes, transcription factor binding and chromatin accessibility. Features were selected using random forest and classifier was designed with supporting vector machine (SVM). A 10-fold cross validation was performed for parameter tuning under several different regression methods and sigmoid functions. The final model acquires a precision of 0.9886 and a recall of 0.9931 on K562 cell lines, with the
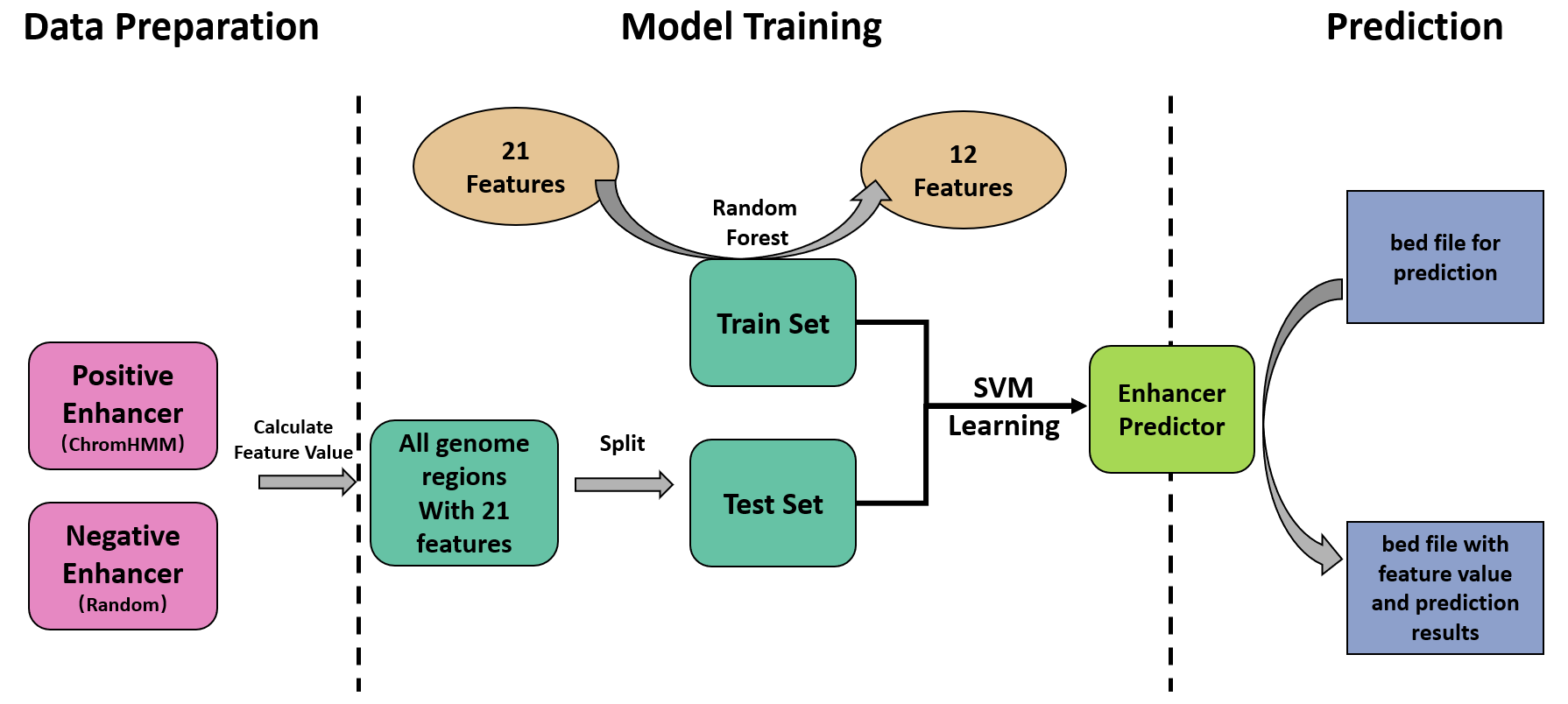
The Broad ChromHMM prediction was adopted as golden standard for enhancer annotation, which is publicly available at UCSC table browser. Regions that were annotated as 'Strong_Enhancer' and has no intersection with TSS regions were retained as true enhancers (positive set, 33393 elements).
Random shuffle was performed to generate non-enhancer genome regions (Negative set) with similar length distribution as the positive set. The data set was then divided into train set and test set randomly.
Multiple gemomic features were collected from public sources, including:
- ChIP-Seq for chromatin modifications: H3K27ac H3K4me1 H3K4me2 H3K9ac H3K4me3 H2az H3K79me2 H3K36me3 H3K9me1 H4K20me1 H3K9me3 H3K27me3
- ChIP-Seq for enzymes for chromation modification: P300 HDAC2A HDAC1 EZH2 HDAC6A SUZ12 CBP
- ChIP-Seq for transcription factor: CTCF
- ATAC-Seq chromatin accessibility
All ChIP-Seq data and corresponding controls were downloaded from ENCODE Broad Histone. ATAC-Seq data was download from GEO with accession number GSE70482.
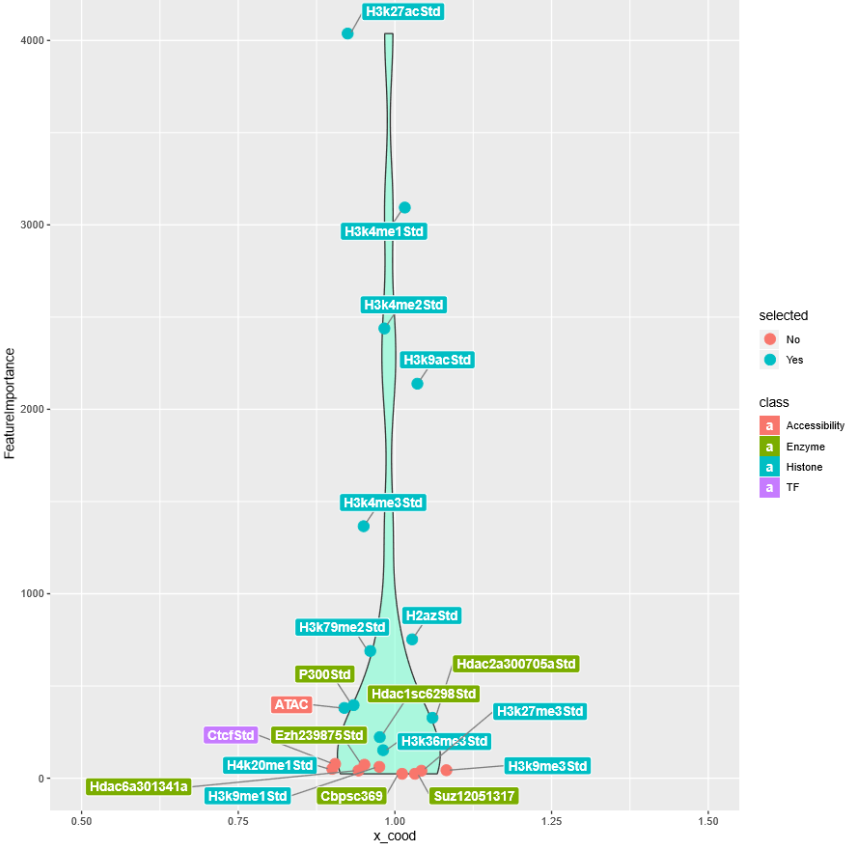
Feature selection was performed with random forest based strategy. A random forest classifier was frist trained with all 21 features to predict enhancer potentials. Gini impurity of each decision tree was calculated and avaraged to give the mean impurity of the whole forest. Next, each fearure was excluded respectily to generate 21 forests with 20 features. The mean decrease in forest impurity (mean decrease impurity, MDI) reflected the importance of the excluded feature. 12 Features with MDI > 100 were retained.
SVM was ultilized as classifier for enhancer prediction. Two different regression methods (C-SVM, Nu-SVM) and four different sigmoid functions (Linear, Polynomial, Radial, Sigmoid) was tested for best performance. A 10-fold cross validation was performed for parameter tuning. The final model acquires a precision of 0.9886 and a recall of 0.9931 on the test set, with the
| Model2 | Truth_Pos | Truth_Neg |
|---|---|---|
| Prediction_True | 16312 | 177 |
| Prediction_False | 114 | 16249 |
Trained model was saved in RData format and could be used with a single integrated script EnhancerPredictor.R. The most simple command to run Enhancer Predictor:
Rscript EnhancerPredictor.R input.bed output.bed
where input.bed is the genome regions to be perform enhancer prediction, saved in bed format. And output.bed is the specified the output file.