Analysis pipeline for MeRIP-seq
 |
 |
Schematic diagram of the MeRIP-seq protocol |
Data analysis workflow |
由于m6A-seq数据分析的原理与过程和ChIP-seq十分相似,所以这里略过前面的质控,简单说明比对和peak calling步骤,具体内容可以参考ChIP-seq分析流程
目前已知有100多种RNA修饰,涉及到mRNAs、tRNAs、rRNAs、small nuclear RNA (snRNAs) 以及 small nucleolar RNAs (snoRNAs)等。其中甲基化修饰是一种非常广泛的修饰,N6-methyl adenosine (m6A)是真核生物mRNA上最为广泛的甲基化修饰之一,并存在于多种多样的物种中。
腺嘌呤可以被编码器METTL3、METTL14和WTAP及其他组分组成的甲基转移酶复合体甲基化,甲基化的腺嘌呤可以被读码器(目前发现m6A读码器主要有四个,定位于细胞核内的YTHDC1以及定位在细胞质中的YTHDF1、YTHDF2、YTHDF3、YTHDC2)识别,同时m6A可以被擦除器FTO和ALKBH5这两个去甲基化酶催化去甲基化。
在哺乳动物mRNA中,m6A修饰存在于7000多个基因中,保守基序为RRACH (R = G, A; H = A, C, U)。m6A修饰富集在mRNA终止密码子附近。
比对参考基因组 目录
在 ChIP-seq 中一般用 BWA 或者 Bowtie 进行完全比对就可以了,但是在 MeRIP-seq 中,由于分析的 RNA ,那么就存在可变剪切,对于存在可变剪切的 mapping 用 Tophat 或者 Tophat 的升级工具 HISAT2 更合适
# build reference index
## <1> build genome index
$ bowtie2-build hg19.fa hg19
## <2> build transcriptome index
$ tophat -p 8 -G hg19.gtf --transcriptome-index=Ref/hg19/hg19_trans/know hg19
# mapping
$ tophat -p 8 --transcriptome-index=Ref/hg19/hg19_trans/know -o outdir hg19 reads1_1.fastq reads1_2.fastq
# Only uniquely mapped reads with mapping quality score ≥20 were kept for the subsequent analysis for each sample
$ samtools view -q 20 -O bam -o outdir/accepted_hits.highQual.bam outdir/accepted_hits.bam
Tophat参数
- -p Number of threads to use
- -G Supply TopHat with a set of gene model annotations and/or known transcripts, as a GTF 2.2 or GFF3 formatted file.
- --transcriptome-index TopHat should be first run with the -G/--GTF option together with the --transcriptome-index option pointing to a directory and a name prefix which will indicate where the transcriptome data files will be stored. Then subsequent TopHat runs using the same --transcriptome-index option value will directly use the transcriptome data created in the first run (no -G option needed after the first run).
- -o Sets the name of the directory in which TopHat will write all of its output
samtools view 参数
- -q only include reads with mapping quality >= INT [0]
# build reference index
## using the python scripts included in the HISAT2 package, extract splice-site and exon information from the gene
annotation fle
$ extract_splice_sites.py chrX_data/genes/chrX.gtf >chrX.ss
$ extract_exons.py chrX_data/genes/chrX.gtf >chrX.exon
## build a HISAT2 index
$ hisat2-build --ss chrX.ss --exon chrX.exon chrX_data/genome/chrX.fa chrX_tran
# mapping
$ hisat2 -p 10 --dta -x chrX_tran -1 reads1_1.fastq -2 reads1_2.fastq | samtools sort -@ 8 -O bam -o reads1.sort.bam 1>map.log 2>&1
Usage: hisat2 [options]* -x <ht2-idx> {-1 <m1> -2 <m2> | -U <r>} [-S <sam>]
- -p Number of threads to use
- --dta reports alignments tailored for transcript assemblers
- -x Hisat2 index
- -1 The 1st input fastq file of paired-end reads
- -2 The 2nd input fastq file of paired-end reads
- -S File for SAM output (default: stdout)
Peak calling 目录
参考ChIP-seq分析流程中的peak calling过程
peakranger ccat --format bam SRR1042594.sorted.bam SRR1042593.sorted.bam \
Xu_MUT_rep1_ccat_report --report --gene_annot_file hg19refGene.txt -q 0.05 -t 4
Peaks注释 目录
哈佛刘小乐实验室出品的软件,可以跟MACS软件call到的peaks文件无缝连接,实现peaks的注释以及可视化分析
CEAS需要三种输入文件:
- Gene annotation table file (sqlite3)
可以到CEAS官网上下载:http://liulab.dfci.harvard.edu/CEAS/src/hg18.refGene.gz ,也可以自己构建:到UCSC上下载,然后用
build_genomeBG脚本转换成split3格式
- BED file with ChIP regions (TXT)
需要包含chromosomes, start, and end locations,这样文件可以由 peak-caller (如MACS2)得到
- WIG file with ChiP enrichment signal (TXT)
如何得到wig文件可以参考samtools操作指南:以WIG文件输出测序深度
CEAS的使用方法很简单:
ceas --name=H3K36me3_ceas --pf-res=20 --gn-group-names='Top 10%,Bottom 10%' \
-g hg19.refGene -b ../paper_results/GSM1278641_Xu_MUT_rep1_BAF155_MUT.peaks.bed \
-w ../rawData/SRR1042593.wig
- --name Experiment name. This will be used to name the output files.
- --pf-res Wig profiling resolution, DEFAULT: 50bp
- --gn-group-names The names of the gene groups in --gn-groups. The gene group names are separated by commas. (eg, --gn-group-names='top 10%,bottom 10%').
- -g Gene annotation table
- -b BED file of ChIP regions
- -w WIG file for either wig profiling or genome background annotation.
Motif识别 目录
安装旧版本的HOMER比较复杂,因为旧版依赖于调用其他几个工具:
- blat
- Ghostscript
- weblogo Does NOT work with version 3.0!!!!
新版HOMER安装很简单,主要是通过configureHomer.pl脚本来安装和管理HOMER
cd ~/biosoft
mkdir homer && cd homer
wget http://homer.salk.edu/homer/configureHomer.pl
# Installing the basic HOMER software
perl configureHomer.pl -install
# Download the hg19 version of the human genome
perl configureHomer.pl -install hg19
安装好后可以进行 Motif Identification
# 提取对应的列给HOMER作为输入文件
# change
# chr1 1454086 1454256 MACS_peak_1 59.88
#to
# MACS_peak_1 chr1 1454086 1454256 +
$ awk '{print $4"\t"$1"\t"$2"\t"$3"\t+"}' macs_peaks.bed >homer_peaks.bed
# MeRIP-seq 中 motif 的长度为6个 nt
$ findMotifsGenome.pl homer_peaks.bed hg19 motifDir -size 200 -len 8,10,12
# 自己指定background sequences,用bedtools shuffle构造随机的suffling peaks
$ bedtools shuffle -i peaks.bed -g <GENOME> >peaks_shuffle.bed
# 用参数"-bg"指定background sequences
$ findMotifsGenome.pl homer_peaks.bed hg19 motifDir -bg peaks_shuffle.bed -size 200 -len 8,10,12
Usage: findMotifsGenome.pl <pos file> <genome> <output directory> [additional options]
注意:
<genome>参数只需要写出genome的序号,不需要写出具体路径bedtools shuffle中的genome文件的格式要求:For example, Human (hg19): chr1 249250621 chr2 243199373 ... chr18_gl000207_random 4262可以使用 UCSC Genome Browser's MySQL database 来获取 chromosome sizes 信息并构建genome文件
mysql --user=genome --host=genome-mysql.cse.ucsc.edu -A -e "select chrom, size from hg19.chromInfo" >hg19.genome
最后得到的文件夹里面有一个详细的网页版报告
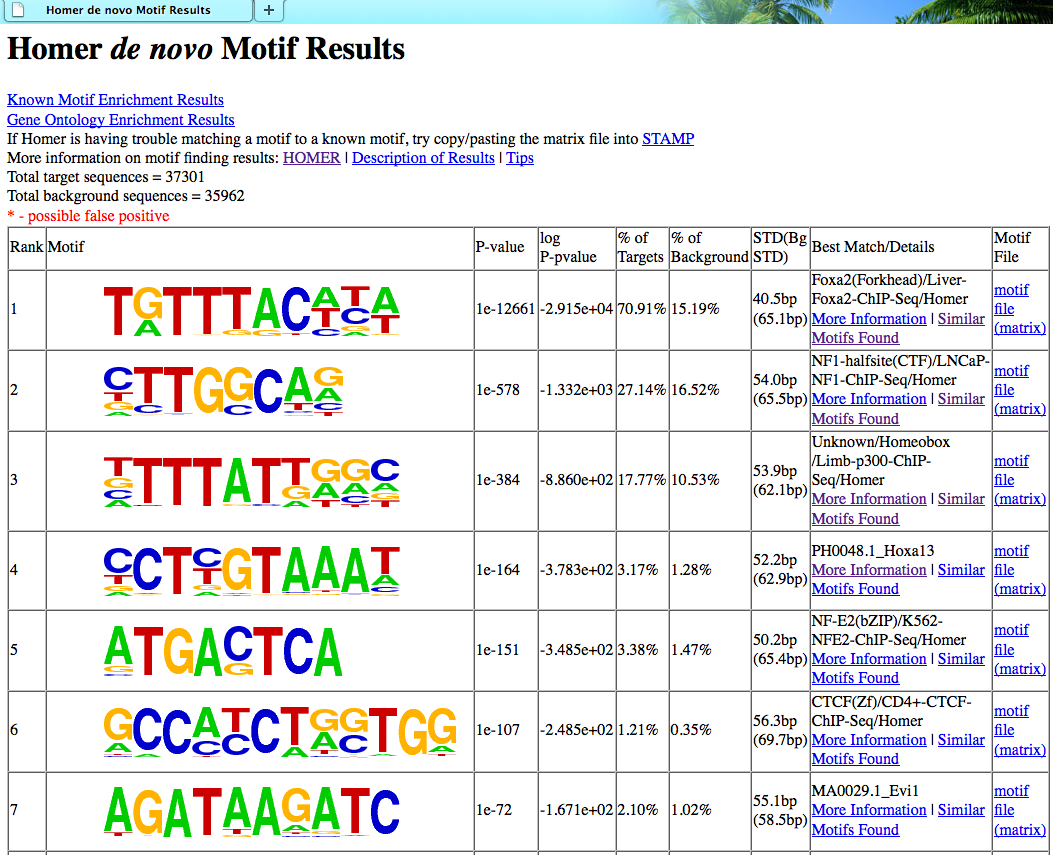
算法原理 目录



操作 目录
下载安装MEME
cd ~/biosoft
mkdir MEMEsuite && cd MEMEsuite
## http://meme-suite.org/doc/download.html
wget http://meme-suite.org/meme-software/4.11.2/meme_4.11.2_1.tar.gz
tar zxvf meme_4.11.2_1.tar.gz
cd meme_4.11.2/
./configure --prefix=$HOME/my-bin/meme --with-url="http://meme-suite.org"
make
make install
# 先提取peaks区域所对应的序列
bedtools getfasta -fi input.fasta -bed input.bed -fo output.fasta
# Motif identification
meme output.fasta -dna -mod oops -pal
Usage: meme <dataset> [optional arguments]
- < dataset > File containing sequences in FASTA format
- -dna Sequences use DNA alphabet
- -mod Distribution of motifs,3 options: oops | zoops | anr
- -pal Force palindromes (requires -dna)
Differential binding analysis 目录
当ChIP-seq数据中有多分组,多样本以及多个重复时,需要进行样本间peaks的merge,先找出各组中至少出现一次(overlap)的peaks作为consensus peakset,然后对不同分组的consensus peakset进行merge
bedtools intersect -a Mcf7H3k27acUcdAlnRep1_peaks.filtered.bed -b Mcf7H3k27acUcdAlnRep2_peaks.filtered.bed -wa | cut -f1-3 | sort | uniq > Mcf7Rep1_peaks.bed
bedtools intersect -a Mcf7H3k27acUcdAlnRep1_peaks.filtered.bed -b Mcf7H3k27acUcdAlnRep2_peaks.filtered.bed -wb | cut -f1-3 | sort | uniq > Mcf7Rep2_peaks.bed
bedtools intersect -a Panc1H3k27acUcdAlnRep1_peaks.filtered.bed -b Panc1H3k27acUcdAlnRep2_peaks.filtered.bed -wa | cut -f1-3 | sort | uniq > Panc1Rep1_peaks.bed
bedtools intersect -a Panc1H3k27acUcdAlnRep1_peaks.filtered.bed -b Panc1H3k27acUcdAlnRep2_peaks.filtered.bed -wb | cut -f1-3 | sort | uniq > Panc1Rep2_peaks.bed
rm *filtered*
cat *bed | sort -k1,1 -k2,2n | bedtools merge > merge.bed
用bedtools
# Make a bed file adding peak id as the fourth colum
$ awk '{$3=$3"\t""peak_"NR}1' OFS="\t" merge.bed > bed_for_multicov.bed
# 输入的bam文件要提前做好index,可同时提供多个bam文件
$ samtools sort -@ 8 -O BAM -o input1.sort.bam input1.bam
$ samtools index -@ 8 input1.sort.bam input1.sort.bam.bai # 注意:生成的索引文件的文件名必须为在原bam文件名后追加".bai",否则bedtools multicov无法识别
$ bedtools multicov -bams input1.bam input2.bam ... -bed bed_for_multicov.bed > counts_multicov.txt
- NR 表示awk开始执行程序后所读取的数据行数
- OFS Out of Field Separator,输出字段分隔符
用featureCounts (subread工具包中的组件)
# Make a saf(simplified annotation format) file for featureCount in the subread package,
shown below:
GeneID Chr Start End Strand
497097 chr1 3204563 3207049 -
497097 chr1 3411783 3411982 -
497097 chr1 3660633 3661579 -
...
$ awk -F "\t" '{$1="peak_"NR FS$1;$4=$4FS"."}1' merge.bed > subread.saf
$ featureCounts -T 4 -a subread.saf -F SAF -o counts_subread.txt ../../data/*bam
Usage: featureCounts [options] -a <annotation_file> -o <output_file> input_file1 [input_file2] ...
- -a Name of an annotation file
- -F Specify format of the provided annotation file. Acceptable formats include 'GTF' (or compatible GFF format) and 'SAF'. 'GTF' by default
- -o Name of the output file including read counts
- -T Number of the threads
- 对 contrast 构建一个 counts 矩阵,第一组的每个样本占据一列,紧接着的是第二组的样本,也是每个样本一列。
# 做好表达矩阵
count_table<-read.delim("count_multicov.txt",header=F)
count_matrix<-as.matrix(count_table[,c(-1,-2,-3,-4)])
rownames(count_matrix)<-count_table$V4
# 做好分组因子
group_list<-factor(c("control","control","control","treat","treat","treat"))
colData <- data.frame(row.names=colnames(count_matrix), group_list=group_list)
# 构建 DESeqDataSet 对象
dds <- DESeqDataSetFromMatrix(countData = count_matrix,
colData = colData,
design = ~ group_list)
- 接着计算每个样本的 library size 用于后续的标准化。library size 等于落在该样本所有peaks上的reads的总数,即 counts 矩阵中每列的加和。如果bFullLibrarySize设为TRUE,则会使用library的总reads数(根据原BAM/BED文件统计获得)。然后使用
estimateDispersions估计统计分布,需要将参数fitType设为'local'
dds<-estimateSizeFactors(dds)
dds<-estimateDispersions(dds,fitType="local")
- 对负二项分布进行显著性检验(Negative Binomial GLM fitting and Wald statistics)
dds <- nbinomWaldTest(dds)
res <- results(dds)
res
## log2 fold change (MLE): condition treated vs untreated
## Wald test p-value: condition treated vs untreated
## DataFrame with 9921 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue
## <numeric> <numeric> <numeric> <numeric> <numeric>
## FBgn0000008 95.144292 0.002276428 0.2237292 0.01017493 0.99188172
## FBgn0000014 1.056523 -0.495113878 2.1431096 -0.23102593 0.81729466
## FBgn0000017 4352.553569 -0.239918945 0.1263378 -1.89902705 0.05756092
## FBgn0000018 418.610484 -0.104673913 0.1484903 -0.70492106 0.48085936
## FBgn0000024 6.406200 0.210848562 0.6895923 0.30575830 0.75978868
## ... ... ... ... ... ...
## FBgn0261570 3208.388610 0.29553289 0.1273514 2.32061001 0.0203079
## FBgn0261572 6.197188 -0.95882276 0.7753130 -1.23669125 0.2162017
## FBgn0261573 2240.979511 0.01271946 0.1133028 0.11226079 0.9106166
## FBgn0261574 4857.680373 0.01539243 0.1925619 0.07993497 0.9362890
## FBgn0261575 10.682520 0.16356865 0.9308661 0.17571663 0.8605166
## padj
## <numeric>
## FBgn0000008 0.9972093
## FBgn0000014 NA
## FBgn0000017 0.2880108
## FBgn0000018 0.8268644
## FBgn0000024 0.9435005
## ... ...
## FBgn0261570 0.1442486
## FBgn0261572 0.6078453
## FBgn0261573 0.9826550
## FBgn0261574 0.9881787
## FBgn0261575 0.9679223
若存在多个分组需要进行两两比较,则需要提取指定的两个分组之间的比较结果
## 提取你想要的差异分析结果,我们这里是treated组对untreated组进行比较
res <- results(dds2, contrast=c("group_list","treated","untreated"))
resOrdered <- res[order(res$padj),]
resOrdered=as.data.frame(resOrdered)
注意两个概念:
Full library size: bam文件中reads的总数
Effective library size: 落在peaks区域的reads的总数
DESeq2中默认使用 Full library size bam (bFullLibrarySize =TRUE),在ChIP-seq中使用 Effective library size 更合适,所有应该设置为bFullLibrarySize =FALSE
在Differential binding analysis: 2. Preparing ChIP-seq count table得到的bed_for_multicov.bed中找DiffBind peaks的bed格式信息
# 用R进行取交集操作
merge_bed<-read.delim("m6A_seq/CallPeak/bed_for_multicov.bed",header=F)
peaks_diffbind<-read.delim("m6A_seq/CallPeak/res_diffBind.txt",header=T)
peaks_diffbind_bed<-merge_bed[merge_bed$V4 %in% rownames(peaks_diffbind),]
# 保存文件
write.table(peaks_diffbind_bed,"m6A_seq/CallPeak/peaks_diffBind.bed",sep="\t",col.names=F,row.names=F,quote=F)
接着,用基因组注释文件GFF/GTF注释peaks
$ bedtools intersect -wa -wb -a m6A_seq/CallPeak/peaks_diffBind.bed -b Ref/mm10/mm10_trans/Mus_musculus.GRCm38.91.gtf >m6A_seq/CallPeak/peaks_diffbind.anno.bed
$ head m6A_seq/CallPeak/peaks_diffbind.anno.bed
## 1 7120312 7120526 peak_9 1 ensembl_havana exon 7120194 7120615 . + . gene_id "ENSMUSG00000051285"; gene_version "17"; transcript_id "ENSMUST00000061280"; transcript_version "16"; exon_number "2"; gene_name "Pcmtd1"; gene_source "ensembl_havana"; gene_biotype "protein_coding"; havana_gene "OTTMUSG00000043373"; havana_gene_version "5"; transcript_name "Pcmtd1-201"; transcript_source "ensembl_havana"; transcript_biotype "protein_coding"; tag "CCDS"; ccds_id "CCDS35508"; havana_transcript "OTTMUST00000113805"; havana_transcript_version "2"; exon_id "ENSMUSE00000553965"; exon_version "2"; tag "basic"; transcript_support_level "1";
## 1 7120312 7120526 peak_9 1 ensembl_havana CDS 7120309 7120615 . + 0 gene_id "ENSMUSG00000051285"; gene_version "17"; transcript_id "ENSMUST00000061280"; transcript_version "16"; exon_number "2"; gene_name "Pcmtd1"; gene_source "ensembl_havana"; gene_biotype "protein_coding"; havana_gene "OTTMUSG00000043373"; havana_gene_version "5"; transcript_name "Pcmtd1-201"; transcript_source "ensembl_havana"; transcript_biotype "protein_coding"; tag "CCDS"; ccds_id "CCDS35508"; havana_transcript "OTTMUST00000113805"; havana_transcript_version "2"; protein_id "ENSMUSP00000059261"; protein_version "9"; tag "basic"; transcript_support_level "1";
## 1 7120312 7120526 peak_9 1 ensembl_havana gene 7088920 7173628 . + . gene_id "ENSMUSG00000051285"; gene_version "17"; gene_name "Pcmtd1"; gene_source "ensembl_havana"; gene_biotype "protein_coding"; havana_gene "OTTMUSG00000043373"; havana_gene_version "5";
## 1 7120312 7120526 peak_9 1 ensembl_havana transcript 7088920 7173628 . + . gene_id "ENSMUSG00000051285"; gene_version "17"; transcript_id "ENSMUST00000061280"; transcript_version "16"; gene_name "Pcmtd1"; gene_source "ensembl_havana"; gene_biotype "protein_coding"; havana_gene "OTTMUSG00000043373"; havana_gene_version "5"; transcript_name "Pcmtd1-201"; transcript_source "ensembl_havana"; transcript_biotype "protein_coding"; tag "CCDS"; ccds_id "CCDS35508"; havana_transcript "OTTMUST00000113805"; havana_transcript_version "2"; tag "basic"; transcript_support_level "1";
## 1 7120312 7120526 peak_9 1 havana transcript 7088930 7169598 . + . gene_id "ENSMUSG00000051285"; gene_version "17"; transcript_id "ENSMUST00000182114"; transcript_version "7"; gene_name "Pcmtd1"; gene_source "ensembl_havana"; gene_biotype "protein_coding"; havana_gene "OTTMUSG00000043373"; havana_gene_version "5"; transcript_name "Pcmtd1-202"; transcript_source "havana"; transcript_biotype "protein_coding"; havana_transcript "OTTMUST00000113813"; havana_transcript_version "2"; tag "cds_end_NF"; tag "mRNA_end_NF"; transcript_support_level "1";
peaks注释基因的富集分析 目录
clusterProfiler: GO enrichment analysis 目录
多数人进行GO富集分析时喜欢使用DAVID,但是由于DAVID的最新版本是在2016年更新的,数据并不是最新的,所以不推荐使用DAVID。
推荐使用bioconductor包clusterProfiler
准备geneList
从DiffBind peaks annotation中获得的peaks_diffbind.anno.bed文件中提取geneID
$ cut -f 13 m6A_seq/CallPeak/peaks_diffbind.anno.bed | \
awk 'BEGIN{FS=";"} {print $1}' | \
perl -ane 'chomp;$F[1]=~ s/\"//g;print "$F[1]\n"' | sort | uniq >m6A_seq/CallPeak/peaks_diffbind_geneList.txt
# 先要载入geneList,变量类型为vector
x <- c("GPX3", "GLRX", "LBP", "CRYAB", "DEFB1", "HCLS1", "SOD2", "HSPA2",
"ORM1", "IGFBP1", "PTHLH", "GPC3", "IGFBP3","TOB1", "MITF", "NDRG1",
"NR1H4", "FGFR3", "PVR", "IL6", "PTPRM", "ERBB2", "NID2", "LAMB1",
"COMP", "PLS3", "MCAM", "SPP1", "LAMC1", "COL4A2", "COL4A1", "MYOC",
"ANXA4", "TFPI2", "CST6", "SLPI", "TIMP2", "CPM", "GGT1", "NNMT",
"MAL", "EEF1A2", "HGD", "TCN2", "CDA", "PCCA", "CRYM", "PDXK",
"STC1", "WARS", "HMOX1", "FXYD2", "RBP4", "SLC6A12", "KDELR3", "ITM2B")
# ID转换,转换需要的信息来自于对应物种的数据包(orgDB),所以使用前需要提前安装好数据包
eg = bitr(x, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
## ID转换支持的ID类型可以通过以下方式查看
keytypes(org.Hs.eg.db)
## 当ID转换涉及KEGG ID时需要使用特殊的函数bitr_kegg,只能在kegg,ncbi-geneid,ncbi-proteinid和uniprot之间进行转换
eg2np <- bitr_kegg(hg, fromType='kegg', toType='ncbi-proteinid', organism='hsa')
GO over-representation test
ego <- enrichGO(gene = gene.df$ENSEMBL,
OrgDb = org.Hs.eg.db,
keytype = 'ENSEMBL',
ont = "CC",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
readable = TRUE)
参数说明:
- gene: a vector of entrez gene id
- OrgDb: OrgDb
- keytype: keytype of input gene
- ont: One of "MF", "BP", and "CC" subontologies
- pvalueCutoff: Cutoff value of pvalue
- pAdjustMethod: one of "holm", "hochberg", "hommel", "bonferroni", "BH", "BY", "fdr", "none"
- qvalueCutoff: qvalue cutoff
- readable: whether mapping gene ID to gene Name
head(ego)
## ID Description GeneRatio
## GO:0005819 GO:0005819 spindle 25/198
## GO:0000779 GO:0000779 condensed chromosome, centromeric region 15/198
## GO:0000775 GO:0000775 chromosome, centromeric region 18/198
## GO:0000776 GO:0000776 kinetochore 15/198
## GO:0000793 GO:0000793 condensed chromosome 18/198
## GO:0005876 GO:0005876 spindle microtubule 10/198
## BgRatio pvalue p.adjust qvalue
## GO:0005819 238/11745 2.090374e-13 5.518588e-11 4.950886e-11
## GO:0000779 90/11745 2.241220e-11 2.958411e-09 2.654077e-09
## GO:0000775 152/11745 7.936845e-11 6.984424e-09 6.265930e-09
## GO:0000776 103/11745 1.649359e-10 8.919109e-09 8.001593e-09
## GO:0000793 159/11745 1.689225e-10 8.919109e-09 8.001593e-09
## GO:0005876 45/11745 2.821374e-09 1.241405e-07 1.113700e-07
## geneID
## GO:0005819 CDCA8/CDC20/KIF23/CENPE/ASPM/DLGAP5/SKA1/NUSAP1/TPX2/TACC3/NEK2/CDK1/MAD2L1/KIF18A/BIRC5/KIF11/TTK/AURKB/PRC1/KIFC1/KIF18B/KIF20A/AURKA/CCNB1/KIF4A
## GO:0000779 CENPE/NDC80/HJURP/SKA1/NEK2/CENPM/CENPN/ERCC6L/MAD2L1/CDT1/BIRC5/NCAPG/AURKB/AURKA/CCNB1
## GO:0000775 CDCA8/CENPE/NDC80/HJURP/SKA1/NEK2/CENPM/CENPN/ERCC6L/MAD2L1/KIF18A/CDT1/BIRC5/TTK/NCAPG/AURKB/AURKA/CCNB1
## GO:0000776 CENPE/NDC80/HJURP/SKA1/NEK2/CENPM/CENPN/ERCC6L/MAD2L1/KIF18A/CDT1/BIRC5/TTK/AURKB/CCNB1
## GO:0000793 CENPE/NDC80/TOP2A/NCAPH/HJURP/SKA1/NEK2/CENPM/CENPN/ERCC6L/MAD2L1/CDT1/BIRC5/NCAPG/AURKB/CHEK1/AURKA/CCNB1
## GO:0005876 SKA1/NUSAP1/CDK1/KIF18A/KIF11/AURKB/PRC1/KIF18B/AURKA/KIF4A
## Count
## GO:0005819 25
## GO:0000779 15
## GO:0000775 18
## GO:0000776 15
## GO:0000793 18
## GO:0005876 10
富集分析结果可视化
# 绘制气泡图
## 可以使用clusterProfiler中提供的绘图函数dotplot
dotplot(ego)
## 也可以使用ggplot2进行自定义绘图
library(ggplot2)
p<-ggplot(ego)+geom_point(aes(x=)

# 绘制GOterm拓扑关系网,依赖topGO和Rgraphviz
library(topGO)
library(Rgraphviz)
# 画出来的拓扑图中节点的文字太小,看不清,这是一个问题
plotGOgraph(ego)

参考资料:
(1) Zhang C, Chen Y, Sun B, et al. m(6)A modulates haematopoietic stem and progenitor cell specification[J]. Nature, 2017, 549(7671):273.
(2) BIG科研 | 细胞质内的m6A结合蛋白YTHDF3促进mRNA的翻译
(3) Pertea M, Kim D, Pertea G, et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie, and Ballgown[J]. Nature Protocols, 2016, 11(9):1650.
(5) ChIPseq pipeline on jmzeng1314's github
(6) ChIPseq pipeline on crazyhottommy's github
(7) library size and normalization for ChIP-seq
(8) Bioconductor tutorial: Analyzing RNA-seq data with DESeq2
(9) Bioconductor tutorial: clusterProfiler
(10) 国科大-韩春生《生物信息学应用 - 模序搜索》ppt